Neil H. Riordan, Maria Luisa Hincapié, Isabela Morales, Giselle Fernandez, Nicole Allen, Cindy Leu, Marialaura Madrigal, Jorge Paz Rodriguez, Nelson Navarro
First published: 11 June 2019 https://doi.org/10.1002/sctm.19-0010
Data Availability Statement: The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abstract
Individuals with autism spectrum disorder (ASD) suffer from developmental disabilities that impact communication, behavior, and social interaction. Immune dysregulation and inflammation have been linked to children with ASD, the latter manifesting in serum levels of macrophage‐derived chemokine (MDC) and thymus, and activation‐regulated chemokine (TARC). Mesenchymal stem cells derived from umbilical cord tissue (UC‐MSCs) have immune‐modulatory and anti‐inflammatory properties, and have been safely used to treat a variety of conditions. This study investigated the safety and efficacy of UC‐MSCs administered to children diagnosed with ASD. Efficacy was evaluated with the Autism Treatment Evaluation Checklist (ATEC) and the Childhood Autism Rating Scale (CARS), and with measurements of MDC and TARC serum levels. Twenty subjects received a dose of 36 million intravenous UC‐MSCs every 12 weeks (four times over a 9‐month period), and were followed up at 3 and 12 months after treatment completion. Adverse events related to treatment were mild or moderate and short in duration. The CARS and ATEC scores of eight subjects decreased over the course of treatment, placing them in a lower ASD symptom category when compared with baseline. MDC and TARC inflammatory cytokine levels also decreased for five of these eight subjects. The mean MDC, TARC, ATEC, and CARS values attained their lowest levels 3 months after the last administration. UC‐MSC administration in children with ASD was therefore determined to be safe. Although some signals of efficacy were observed in a small group of children, possible links between inflammation levels and ASD symptoms should be further investigated.
Significance Statement
To the authors’ knowledge, this is the first single‐arm phase I/II clinical trial of repeated dose umbilical cord mesenchymal stem cells administration in children diagnosed with autism spectrum disorder (ASD). Umbilical cord mesenchymal stem cell infusions were safe and generally well tolerated. Forty percent of children showed notable improvements of symptoms as measured by standardized autism diagnosis tools. Whereas other studies have reported links between inflammatory cytokine levels and ASD, this study only observed a possible link in a small group of children, which merits further investigation.
Introduction
Individuals with autism spectrum disorder (ASD) suffer from developmental disabilities that impact communication, behavior, and social interaction. Although the clinical presentation of this disorder varies in the presence and intensity of the signs and symptoms displayed, children with ASD typically present repetitive behavior and speech patterns, as well as deficits in social interactions and verbal/nonverbal communication. Additionally, anxiety, attention‐deficit/hyperactivity disorder, motor impairments (e.g., hypotonia, clumsiness, toe‐walking), sleep disorders (e.g., insomnia), intellectual disability, and gastrointestinal problems (e.g., chronic constipation, diarrhea, abdominal pain) are also associated with ASD1.
The prevalence of autism, which is approximately four times more frequent in boys than girls, has increased in recent years2, causing a significant economic burden in special education, healthcare costs, and parental productivity loss3,4. Current management of the condition is limited to psychological interventions and other alternative therapies (behavioral, cognitive, and speech therapy)5, and management of symptoms with pharmacotherapy (e.g., selective serotonin reuptake inhibitors [SSRIs], antipsychotic medications6 known for causing adverse effects such as extrapyramidal symptoms, sedation, weight gain, among others1,7,8). However, despite the growing number of cases and the financial and social impact of this condition, the benefits of these interventions may be limited, prompting the need for biologic approaches targeting the etiology of ASD at the cellular and molecular level.
Immune dysregulation has been linked to children with ASD, manifesting in the form of altered T‐cell responses9, elevated plasma cytokine levels10, and significantly lower plasma levels of transforming growth factor β‐111, among others12,13. In particular, intestinal immune dysregulation14 and gastrointestinal symptoms have been observed in children with ASD15-19. Furthermore, brain inflammation may be linked to the pathogenesis of neuropsychiatric disorders such as ASD20, as observed through findings that indicate neurological inflammation, including neural fiber formation21, enhanced oxidative stress22, apoptosis23, and high secretion of amyloid protein breakdown products24. The relationship between inflammation and autism was further evidenced in a study by Al‐Ayadhi and Mostafa, in which children with ASD were found to score higher than neurotypical children in measures of macrophage‐derived chemokine (MDC) and thymus and activation‐regulated chemokine (TARC). Additionally, those with severe autism based on the Childhood Autism Rating Scale (CARS) had significantly higher serum levels than those with mild to moderate autism25.
Mesenchymal stem cells (MSCs) have immune‐modulatory and anti‐inflammatory properties and have been safely used in the treatment of a variety of neurological and autoimmune conditions15, 26-33. In particular, MSCs derived from the Wharton’s jelly of umbilical cord tissue (UC‐MSCs) may possess greater immune‐modulatory activity34 and proliferative capacity compared with other MSCs35,36. The rationale for MSC therapy to treat ASD has been discussed over the past decade37,38; our group proposed the use of stem cell therapy to treat ASD in 200739. Some studies to date have demonstrated the safety of treatment that included MSCs40: of note, the results of a study by Sharma et al. showed that the majority (96%) of children with ASD treated with bone marrow‐derived cells including MSCs showed global improvements including behavior patterns (66%), social relationships (90.6%), and speech, language, and communication (78%)41. In another study, children with ASD treated with UC cells, including MSCs, showed significant differences in nonverbal communication and visual, emotional, and intellectual responses, among other measures42.
In this context, the purpose of this study was to analyze the safety and signals of therapeutic effects of a 9‐month intervention of intravenously administered UC‐MSCs in 20 children diagnosed with ASD.
Materials and Methods
Study Design
In this single‐arm phase I/II clinical trial of 20 subjects with ASD, enrolled subjects received one treatment series every 12 weeks for a total of four treatment series over the course of 9 months (treatment phase). Subjects were then followed for 1 year, with evaluations 3 and 12 months after the last treatment (12‐month and 21‐month visits, respectively). Complete medical and psychiatric evaluations, complete blood count, complete metabolic panel, and infectious disease tests, serum cytokine levels (MDC and TARC), and autism‐specific questionnaires (CARS and Autism Treatment Evaluation Checklist [ATEC]) were administered at each time point during the treatment and follow‐up phases.
During the first visit, in week 1, participants were evaluated for safety and efficacy baseline values, and received 36 million UC‐MSCs intravenously over the course of 1 week, in four intravenous infusions of 9 million viable UC‐MSCs in each infusion. Twelve weeks later, at week 13, the subjects received the same dose of UC‐MSCs and were evaluated for safety and efficacy endpoints. This procedure was repeated at week 25 and week 37 after the start of treatment. The total dose received over the course of treatment was 144 million UC‐MSCs (4 × 36 million). In the follow‐up phase, visits occurred at week 49 (12 months after the start of treatment, 3 months after the last dose) and week 89 (21 months after the start of treatment, 12 months after the last dose).
The study was approved by the Panamanian Institutional Review Board (Comité Nacional de Bioética de la Investigación) and registered with the National Institutes of Health U.S. National Library of Medicine database (clinicalTrials.gov identifier NCT02192749). The study was sponsored by Translational Biosciences. All treatments were administered at the Stem Cell Institute in Panama City, Republic of Panama, under protocol number TBS‐UCMSC‐ASD001. Written informed consent was obtained for all study participants and cord donors.
Subject Population
Stem cell therapy‐naïve children aged 6–16 years were considered for this study if they had a prior diagnosis of autism per the Diagnostic and Statistical Manual of Mental Disorders, 4th edition, as confirmed by the Autism Diagnostic Observation Schedule or the Autism Diagnostic Interview—Revised. Eligible candidates were required to be ambulatory, able to sit still for at least 5 minutes, and have adequate vision, hearing, and arm‐hand‐finger coordination (i.e., be able to point), as well as have normal serum lead and mercury levels at screening and no other uncontrolled medical disorders. Children who were premature (<32 weeks gestation) or significantly small for gestational age were excluded from the trial, as were those with intellectual disability, seizure disorders, auto‐immune conditions, or a history of head trauma. Additionally, candidates were not considered eligible if they had recently made or anticipated any changes in their routine treatment or diet.
UC‐MSC Preparation and Culture
UC‐MSCs used in this study were isolated from human UC tissue from voluntarily donated UCs, obtained from normal healthy births after a rigorous screening process. In brief, a standard risk assessment questionnaire was given to the mothers aged 18–35 years old at the time of delivery, and the donor was screened for infectious diseases (including Human Immunodeficiency Virus (HIV) [1+2]‐Ab and HIV [1+2] Ag‐Ab, V.D.R.L., Hepatitis B (HB)sAg and HB‐anti‐core/IgG‐IgM, cytomegalovirus IgM, Hepatitis A Virus‐IgM, Hepatitis C Virus‐Ab, Chagas‐Ab, Human T‐lymphotropic Virus [1+2]‐Ab, and toxoplasmosis IgM [this disease endemic to Panama is routinely scanned as part of standard of care]). The cells used for this study were manufactured by MediStem Panama, a biotechnology laboratory located in the International Science and Technology Park, City of Knowledge, Panama, following good manufacturing and laboratory practices.
UC‐MSCs were obtained through the enzymatic digestion of UC Wharton’s jelly using collagenase 1.67% (Sigma, C9891, Saint Louis, MO, USA) at 37°C. After isolation, cells were expanded up to passage 5 using α‐MEM (Gibco, 32561‐102, Carlsbad, CA, USA) supplemented with 4 mM GlutaMax (Gibco, 35050‐079, Carlsbad, CA, USA) and 10% inactivated fetal bovine serum (Gibco, 16000044, Grand Island, NY, USA). Vials containing only UC‐MSCs were cryopreserved using 6% hydroxyethyl starch (Claris, G/LVP‐5, Ellisbridge, Ahmedabad, India) containing 10% Dimethyl sulfoxide (Sigma, D2650, Irvine, United Kingdom), first cooled in the −80°C ultra‐freezer at approximately 1°C/minute from 25°C to −80°C in a freezing container (Nalgene, 5100‐0001, Rochester, NY, USA), and then plunged directly into the gas phase of liquid nitrogen. They were kept in quarantine until it was confirmed that they met the requirements for viability (before freezing and after thawing), sterility, mycoplasma, endotoxin, characterization, and differentiation, by testing random vials of the same lot. Before each treatment, cell vials were selected according to the number of total viable cells as obtained by quality controls after freezing and thawing (following syringe preparation procedure), to have the dose of cells required by the protocol. After post‐thaw washing, the dose was adjusted to attain the treatment target of 9 million cells per infusion as closely as possible.
Vials were thawed under controlled conditions and prepared into the corresponding treatment dose of 2.25 million cells per milliliter, suspended in a 4‐ml solution (1 ml 5% dextrose and 3 ml sterile 0.85% saline), for a total of 9 million viable cells per infusion. The procedures were done under strict adherence to aseptic technique to ensure sterility of the prepared syringe and following the results of quality control vials to ensure viability of the cells. Each syringe was inspected for the absence of cell clumps, integrity of the containers, and correct volume. Labels were checked to verify traceability against the provided documents of certificate of analysis of the lot and chain of custody. Viability, characterization, and differentiation methodologies were validated both internally and by a third‐party independent laboratory. Cells were counted and viability was measured using flow cytometer with the Guava ViaCount Reagent (MerckMillipore, 4000‐0041, Hayward, CA, USA) from time 0 to 4 hours at room temperature (20–24°C). Once syringes were prepared, the cells were infused in less than 2 hours, as this was determined by a post‐thaw stability study (data not shown) to be an optimal threshold to preserve stability. Only cells with a post‐thaw viability ≥75% (mean viability 86.5%, SD 3.63%, coefficient of variance 4.20%, median 88.0%, minimum 76.8%, maximum 93.6%); negative for aerobes, anaerobes, and mycoplasma; with an endotoxin level ≤ 3.0 EU/ml; ≥95% positive for CD90, CD73, and CD105 cell surface markers; negative for CD34 and CD45 cell surface markers according to the International Society for Cellular Therapy criteria for MSC43; and with the ability to differentiate into adipocytes, chondrocytes, and osteocytes were used clinically.
Study Endpoints
Safety, the primary endpoint of this study, was assessed at six different time points during the study through complete psychiatric and medical evaluations, safety laboratory exams (complete blood count, complete metabolic panel, and infectious disease tests), occurrence of adverse events and serious adverse events, and their relatedness to the study product.
Signals of efficacy were evaluated by parent‐reported outcomes via the CARS and ATEC tools44, in collaboration with the study pediatric psychiatrist, who evaluated the appearance, behavior, mood, speech, and intellectual functioning of the subjects to supplement parental reports. The second set of efficacy measures, MDC and TARC serum levels, were measured using enzyme‐linked immunosorbent assay in duplicate by RayBiotech, Inc. Service division. Optical density was measured to determine average concentration per milliliter.
Statistical Analysis
Data were analyzed using IBM SPSS software version 25. Mean, SD, minimum, and maximum values were calculated for MDC and TARC levels, and CARS and ATEC scores at six different time points: week 1 (T1, baseline), week 13 (T2, second treatment series), week 25 (T3, third treatment series), week 37 (T4, fourth treatment series), week 49 (12‐month visit), and week 89 (21‐month visit). Missing data were analyzed in order to determine whether data were missing completely at random using Little’s MCAR test. An EM algorithm with a maximum of 25 iterations was used to attempt to replace missing values. To determine whether the treatment had a significant therapeutic effect, a test of difference of repeated measures multivariate analysis of variance (MANOVA) was conducted to determine whether there were significant changes in the mean MDC, TARC, ATEC, and CARS values at any of the six time points for participants who had a complete data set. A level of significance of p < .05 was used for all analyses.
Results
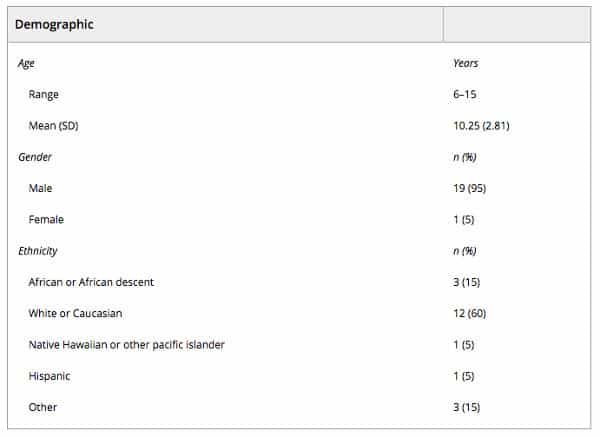
Twenty subjects of diverse ethnicities were enrolled into this study between March 2015 and December 2015. Of these, most (95%) were male, and the average age of enrollees was 10.25 years (Table 1). Average baseline CARS and ATEC scores were 37.48 and 61.10, respectively, and average pretreatment serum MDC and TARC levels were 949.60 and 212.35, respectively. Of the enrolled subjects, 16 completed all four treatment series specified in the study protocol; 296 infusions were administered in total. Subjects received a total dose of 36 million UC‐MSCs at each treatment time point (mean 36.1 million, SD 0.06, coefficient of variance 0.16%, median 36.1, minimum 36.03, maximum 36.16), for a total of 144 million over the course of treatment (mean 144.3 million, SD 0.19, coefficient of variance 0.13%, median 144.3, minimum 144.07, maximum 144.73) for those who completed the treatment series (n = 16). Fifteen subjects were followed to the end of the study period (five did not complete it: two subjects discontinued after receiving two treatment series due to their parents being significantly ill and unable to comply with the study visits, two children discontinued for personal reasons after completing three treatment series, and one was lost to follow‐up after completing the entire treatment phase). Missing data were found to be missing completely at random under Little’s MCAR test (χ2 [131] = 121.60, p = .71). The number of subjects who received treatment and had a fully complete set of efficacy endpoints (all CARS scores, ATEC scores, MDC, and TARC serum levels) at all time points of the study was 10.
Table 1. Demographics of the study population (n = 20)
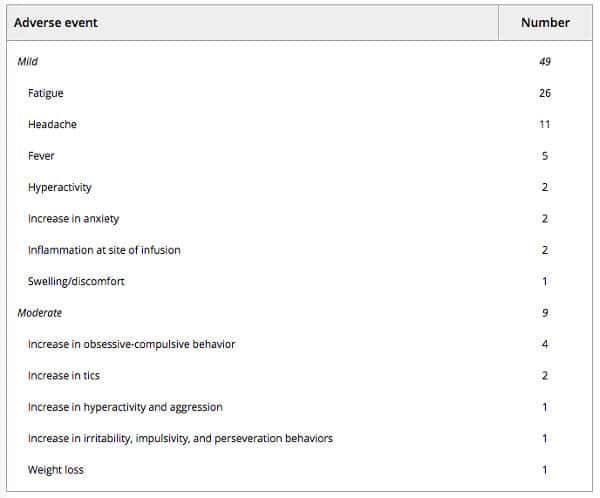
No treatment‐related serious adverse events (SAEs) were observed during the course of this trial. There was one instance of an aggression crisis that required hospitalization in a patient with a documented history of severe aggression prior to entering the study. In total, 133 adverse events were recorded for 296 infusions, of which 58 (19.6%) were considered to be related to treatment with UC‐MSCs (Table 2). Most of the adverse events (AEs) observed during the study (56.4%) were qualified as “not related” or “not likely related” to treatment with UC‐MSCs. Mild inflammation, swelling, and/or redness at the infusion site were reported by two subjects as short in duration and self‐resolved, and were determined to be “definitely related” to treatment. AEs that were considered “possibly related” to treatment included moderate increases in tics, obsessive–compulsive behaviors, and aggression reported by six subjects, as well as mild fatigue, headache, fever, and increase in hyperactivity or anxiety. No clinically significant changes in basic hematologic and chemistry laboratory tests were observed throughout the duration of the study.
Table 2. Number and severity of adverse events related to treatment over the course of treatment, for a total of 296 UC‐MSC intravenous infusions administered
Abbreviation: UC‐MSC, mesenchymal stem cells derived from umbilical cord tissue.
The repeated measures MANOVA (Table 3, n = 10) showed that the mean MDC (p = .003, ηp2 = 0.63), TARC (p = .001, ηp2 = 0.70), ATEC (p = .005, ηp2 = 0.60), and CARS (p < .001, ηp2 = 0.77) values were significantly different at the six different time points.
Table 3. Repeated measures MANOVA of differences in mean MDC, TARC, ATEC, and CARS values (n = 10)

a p < .05.
Abbreviations: ATEC, Autism Treatment Evaluation Checklist; CARS, Childhood Autism Rating Scale; MANOVA, multivariate analysis of variance; MDC, macrophage‐derived chemokine; TARC, thymus and activation‐regulated chemokine.
MDC levels (Fig. 1A, n = 20, 20, 18, 16, 13, and 10 at each time point) remained relatively stable from T1 (mean = 949.60; SD = 165.30) to T3 (mean = 951.33; SD = 200.73), after which a decreasing trend was observed at T4 (mean = 801.23; SD = 383.81) until the 12‐month visit, where the values were halved (mean = 483.46; SD = 343.66) from those measured at T1. Serum MDC levels increased again up to the 21‐month visit (mean = 899.25; SD = 342.78) to levels similar to or lower than those measured at T1.
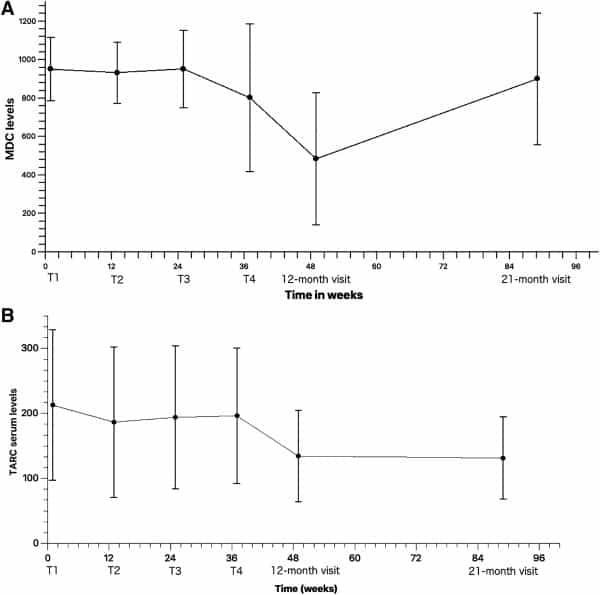
Figure 1.
(A): Mean serum macrophage‐derived chemokine levels at the four treatment points and the 12‐month and 21‐month visits (n = 20, 20, 18, 16, 13, and 10, respectively). (B): Mean serum thymus and activation‐regulated chemokine levels at the four treatment points and the 12‐month and 21‐month visits (n = 20, 20, 18, 16, 13, and 10, respectively).
Although showing a decreasing trend when compared with baseline (mean = 212.35; SD = 115.82), TARC serum levels increased slightly between T2 (mean = 186.21; SD = 115.95), and T4 (mean = 195.93; SD = 104.34), after which a decrease was observed at the 12‐month visit (mean = 134.05; SD = 70.09). TARC levels continued to decrease thereafter, with the lowest results seen at the 21‐month visit (mean = 130.90; SD = 63.32) compared with those measured at baseline (Fig. 1B, n = 20, 20, 18, 16, 13, and 10 at each time point).
Scores for ATEC (Fig. 2A, n = 20, 20, 18, 17, 14, and 14 at each time point) and CARS (Fig. 2B, n = 20, 20, 18, 17, 15, and 14 at each time point) followed a decreasing trend during treatment, with the lowest scores observed at the 12‐month visit (ATEC: mean = 39.14, SD = 22.85; CARS: mean = 31.17, SD = 8.79). The values increased at the 21‐month visit, reaching levels similar to or lower than those observed before treatment.

Figure 2.
(A): Mean Autism Treatment Evaluation Checklist scores at the four treatment points and the 12‐month and 21‐month visits (n = 20, 20, 18, 17, 14, and 14, respectively). (B): Mean Childhood Autism Rating Scale scores at the four treatment points and the 12‐month and 21‐month visits (n = 20, 20, 18, 17, 15, and 14, respectively).
The greatest change or decrease in means was observed for all efficacy variables at the 12‐month visit time point (MDC −465.52; TARC −81.95; ATEC −24.29; CARS −6.83) when compared with baseline (Table 4). At the 12‐month visit, eight subjects (40%) had improvements in their CARS scores that placed them in a lower threshold category of autism symptoms when compared with baseline (Fig. 3). Of these, five (62.5%) improved from CARS scores indicating mild or moderate autism to below the threshold for autism, and three (37.5%) improved from symptoms of severe autism to below the threshold for autism. Additionally, these eight subjects also showed improvements in the ATEC scale that signified drops into lower percentiles at the 12‐month visit compared with baseline; notably, five of them (62.5%) scored in the 10% percentile (<30, mild autism). Five (62.5%) of these subjects also showed a decrease in MDC and TARC levels at the 12‐month visit. Two participants had an improvement in the CARS scale at the 12‐month visit but had higher MDC levels, and three had no category improvements in the CARS scale but had lower MDC and TARC levels.
Table 4. Descriptive statistics of changes in MDC, TARC, ATEC, and CARS values
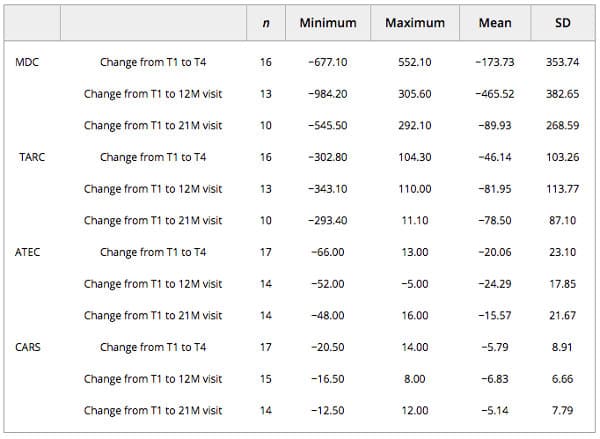
Abbreviations: ATEC, Autism Treatment Evaluation Checklist; CARS, Childhood Autism Rating Scale; MDC, macrophage‐derived chemokine; TARC, thymus and activation‐regulated chemokine.
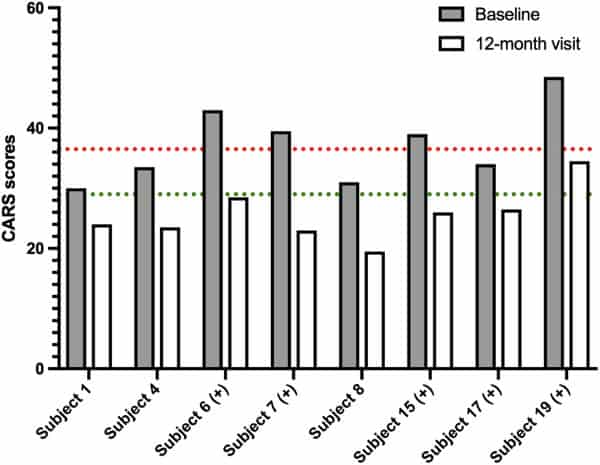
Figure 3
Improvements in Childhood Autism Rating Scale (CARS) scores for eight participants between baseline and the 12‐month visit. Each bar represents the individual CARS score of one participant. Dotted lines correspond to the CARS thresholds for severity (severe at >37 in red, mild between 30 and 36.5 in green, and below autism threshold <30). For five subjects indicated with (+), thymus and activation‐regulated chemokine and macrophage‐derived chemokine mean serum levels also decreased between baseline and the 12‐month visit.
Discussion
To the best of our knowledge, this study was the first to analyze the safety and the effects of repeated, periodic administration of Wharton’s jelly tissue‐derived UC‐MSCs in children diagnosed with ASD, treated over a 9‐month period and followed up for 1 year after the end of treatment.
UC‐MSC administration was safe and well tolerated by children who participated in this trial and no treatment‐related serious adverse events were observed. The adverse events related to treatment were mild or moderate in intensity and short in duration, generally resolving by the end of each treatment visit without the need for medications. Of note, of the observed adverse events, headaches, fever, and fatigue are side effects commonly reported in treatments with MSCs45,46. The observed increase in tics, aggressiveness, and obsessive–compulsive behaviors in some subjects has sometimes been reported in other studies investigating new treatments for ASD, including groups receiving only a placebo47,48. This is perhaps indicative of the sensitiveness of this particular study population to new and unusual situations or changes in routines. Although every effort was made to guarantee the comfort of the subjects in this trial, it would be ideal to conduct further research in environments that minimize stressors and known triggers for patients with ASD to rule out a possible effect of UC‐MSCs in behavioral changes.
Eight subjects showed decreased CARS and ATEC scores over the course of treatment, to the point of changing symptom categories in their respective scales. The scores of three participants of this group, remarkably, changed from the category of severe autism to below the autism threshold on the CARS when the 12‐month visit was compared with baseline. Although five of these eight subjects also presented a decrease in MDC and TARC levels, suggesting a possible link between inflammation levels and ASD symptom manifestations reported by other groups25, we observed that changes in inflammatory markers did not always correlate to response to treatment in all subjects. Periodic psychiatric evaluations showed that the group of children who presented improvements in efficacy variables also manifested increased awareness, and noticeable improvements in social communication (both verbal and expressive) and motor ability, despite causing an increase in anxiety and emotional liability in some of them. The causes for the variability between subjects who showed improvements and those who did not should be further investigated in studies with a more homogeneous subject population, where environmental, molecular, or genetic factors are ruled out or controlled.
Although there is no consensus in the literature regarding optimal dosage or frequency of MSC administration, preclinical studies have reported that repeated doses of MSCs are more beneficial than a single dose for the treatment of other conditions49-51. Repeated dosage has consequently been proposed as a new paradigm in stem cell therapy52,53 and has been applied for other medical conditions15,54. From our previous clinical observations, the therapeutic effect of MSC infusions often declines between 3 and 6 months after administration, likely due to the immune‐evasive properties of MSCs55 that allow them to persist in the body before being eliminated. Therefore, we designed this repeated dose‐study with the intent of maximizing the potential anti‐inflammatory effects of UC‐MSCs by spacing out treatment visits every 12 weeks. The mean of MDC and TARC values and of ATEC and CARS scores (Figs. 1, 2) attained their lowest levels at the 12‐month visit, indicating that the effect of the last UC‐MSC treatment still persisted 3 months after the last administration. Additionally, the means of MDC, CARS, and ATEC experienced an increase in the absence of MSC treatment (between the 12‐month visit and the 21‐month visit), although still remaining below the levels seen at baseline. Interestingly, after a sharp decline between the last treatment and the 12‐month visit, mean TARC levels stayed down until the 21‐month visit. We intend to focus on TARC levels as well as other inflammatory markers after UC‐MSC administration in future studies to confirm this finding.
The major limitation of this study was the small sample size, which impacted the statistical power of the analyses. Although a sample size of 20 subjects was still within FDA guidelines for phase I clinical trials, measurements at all time points were not completed by all participants, due to loss to follow‐up and withdrawal from the study for personal reasons. The data were found to be missing completely at random, but the size of the sample did not allow for EM convergence in the sensitivity analysis, and the MANOVA analysis was only performed with the measurements of 10 participants. Additionally, the small sample size also prevented a statistical quantification of the possible correlation between the lower CARS and ATEC scores and the decrease in MDC and TARC levels, in a subgroup of subjects that responded particularly well to treatment. Another limitation of this study is the lack of a placebo comparison group, which prevents attributing improvements to the treatment, especially considering that a placebo effect in caregivers and investigators has been documented in pediatric ASD randomized clinical trials56. The original CARS version was used for this study rather than the preferred and more updated version CARS‐2; in future studies with a larger population, this second version should be used and completed by clinicians to ensure standardized testing rather than relying on parental observations. Although encouraging, the results of this study should therefore be taken as indicative of trends and signals that should be further explored in larger, double‐blind, placebo‐controlled studies.
Conclusion
The administration of repeated‐dose UC‐MSC infusions is safe and tolerable for patients with ASD. Although this phase I study included a small number of subjects without a placebo arm, the trends observed in this study are indicative of potential therapeutic benefits, reflected in lower CARS and ATEC scores that may be associated with decreases in TARC and MDC levels.
Lessons Learned
Immune dysregulation and inflammation have been linked to children with ASD, and MSCs have anti‐inflammatory and immune‐regulating properties that made them an interesting option to treat ASD symptoms. With this study, we have established that repeated infusions with UC MSCs are safe, resulting only in mild or moderate adverse events short in duration and no serious adverse events related to treatment. Although the sample size was limited, we were able to detect improvements in ASD symptoms and in inflammatory cytokine levels in a small group of children. We hope these results will pave the way for more investigations with UC stem cells and will help establish a novel paradigm for addressing inflammatory‐associated neurological conditions with a safe biological therapy.
Acknowledgments
This study was supported by Translational Biosciences, a department of MediStem Panama. The sponsor had the responsibility of initiating, managing, and financing of this research project. We thank Ms. Dorita Avila for her assistance in the preparation, edition, and revision of this article; Dr. Hernán Hernández for his expertise in obtaining peripheral venous access in challenging subjects; Ms. Erika Chandler for the management of the samples for cytokine analysis; Precision Consulting for the statistical analysis, interpretation of the data, and for providing the first draft of figures for the article; and MediStem Panama and Stem Cell Institute staff for their laboratory, medical, and administrative assistance.
Author Contributions
N.H.R.: conceived the original idea, designed the study protocol, searched the literature, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; M.L.H.: performed all the psychiatric evaluations, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; I.M.: supervised the conduct of the study, project manager for the study, wrote the first draft of this manuscript, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; G.F.: main clinical trial coordinator, performed all regulatory communications, collected all the study data with the aid of C.L., wrote the first draft of this manuscript, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; N.A.: led data management, raw data summaries, clinicalTrials.gov updates, wrote the first draft of this manuscript, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; C.L.: provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; M.M.: coordinated the collection and results of cytokine levels, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; J.P.R.: conceived the original idea, designed the study protocol, searched the literature, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript; N.N.: supervised the conduct of the study, provided critical feedback, helped shape the research analysis and manuscript, read and approved the final manuscript.
Disclosure of Potential Conflicts of Interest
N.H.R. and J.P.R. declared leadership position, patent holder, and shareholders of MediStem Panama and the Stem Cell Institute. M.L.H. declared research funding as sub-investigator for Stem Cell Institute. I.M., N.A. declared leadership position with MediStem Panama. G.F., C.L. declared leadership position with Stem Cell Institute. M.M. declared leadership position and stock ownership with MediStem Panama. N.N. declared research funding from MediStem Panama.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
1 Sanchack, KE, Thomas, CA. Autism spectrum disorder: Primary care principles. Am Fam Phys 2016; 94: 972– 979.
2 Rice, CE, Rosanoff, M, Dawson, G et al. Evaluating changes in the prevalence of the autism spectrum disorders (ASDs). Public Health Rev 2012; 34: 1– 22.
3 Lavelle, TA, Weinstein, MC, Newhouse, JP et al. Economic burden of childhood autism spectrum disorders. Pediatrics 2014; 133: e520– e529.
4 Buescher, AV, Cidav, Z, Knapp, M et al. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr 2014; 168: 721– 728.
5 Bhat, S, Acharya, UR, Adeli, H et al. Autism: Cause factors, early diagnosis and therapies. Rev Neurosci 2014; 25: 841– 850.
6 Park, SY, Cervesi, C, Galling, B et al. Antipsychotic use trends in youth with autism spectrum disorder and/or intellectual disability: A meta‐analysis. J Am Acad Child Adolesc Psychiatry 2016; 55: 456.e454– 468.e454.
7 Sharma, SR, Gonda, X, Tarazi, FI. Autism spectrum disorder: Classification, diagnosis and therapy. Pharmacol Ther 2018; 190: 91– 104.
8 Golnik, AE, Ireland, M. Complementary alternative medicine for children with autism: A physician survey. J Autism Dev Disord 2009; 39: 996– 1005.
9 Ashwood, P, Krakowiak, P, Hertz‐Picciotto, I et al. Altered T cell responses in children with autism. Brain Behav Immun 2011; 25: 840– 849.
10 Ashwood, P, Krakowiak, P, Hertz‐Picciotto, I et al. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun 2011; 25: 40– 45.
11 Ashwood, P, Enstrom, A, Krakowiak, P et al. Decreased transforming growth factor beta1 in autism: A potential link between immune dysregulation and impairment in clinical behavioral outcomes. J Neuroimmunol 2008; 204: 149– 153.
12 Ashwood, P, Wills, S, Van de Water, J. The immune response in autism: A new frontier for autism research. J Leukoc Biol 2006; 80: 1– 15.
13 Careaga, M, Van de Water, J, Ashwood, P. Immune dysfunction in autism: A pathway to treatment. Neurotherapeutics 2010; 7: 283– 292.
14 Ashwood, P, Anthony, A, Pellicer, AA et al. Intestinal lymphocyte populations in children with regressive autism: Evidence for extensive mucosal immunopathology. J Clin Immunol 2003; 23: 504– 517.
15 Adams, JB, Johansen, LJ, Powell, LD et al. Gastrointestinal flora and gastrointestinal status in children with autism—Comparisons to typical children and correlation with autism severity. BMC Gastroenterol 2011; 11: 22.
16 Buie, T, Campbell, DB, Fuchs, GJ et al. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: A consensus report. Pediatrics 2010; 125: S1– S18.
17 Horvath, K, Perman, JA. Autism and gastrointestinal symptoms. Curr Gastroenterol Rep 2002; 4: 251– 258.
18 Molloy, CA, Manning‐Courtney, P. Prevalence of chronic gastrointestinal symptoms in children with autism and autistic spectrum disorders. Autism Int J Res Pract 2003; 7: 165– 171.
19 Nikolov, RN, Bearss, KE, Lettinga, J et al. Gastrointestinal symptoms in a sample of children with pervasive developmental disorders. J Autism Dev Disord 2009; 39: 405– 413.
20 Theoharides, TC, Asadi, S, Patel, AB. Focal brain inflammation and autism. J Neuroinflammation 2013; 10: 46.
21 Bode, MK, Mattila, ML, Kiviniemi, V et al. White matter in autism spectrum disorders—Evidence of impaired fiber formation. Acta Radiol 2011; 52: 1169– 1174.
22 Essa, MM, Guillemin, GJ, Waly, MI et al. Increased markers of oxidative stress in autistic children of the Sultanate of Oman. Biol Trace Elem Res 2012; 147: 25– 27.
23 Dong, D, Zielke, HR, Yeh, D et al. Cellular stress and apoptosis contribute to the pathogenesis of autism spectrum disorder. Autism Res 2018; 11: 1076– 1090.
24 Ray, B, Long, JM, Sokol, DK et al. Increased secreted amyloid precursor protein‐alpha (sAPPalpha) in severe autism: Proposal of a specific, anabolic pathway and putative biomarker. PLoS One 2011; 6:e20405.
25 Al‐Ayadhi, LY, Mostafa, GA. Elevated serum levels of macrophage‐derived chemokine and thymus and activation‐regulated chemokine in autistic children. J Neuroinflammation 2013; 10: 72.
26 Shi, Y, Wang, Y, Li, Q et al. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat Rev Nephrol 2018; 14: 493– 507.
27 Hu, J, Yu, X, Wang, Z et al. Long term effects of the implantation of Wharton’s jelly‐derived mesenchymal stem cells from the umbilical cord for newly‐onset type 1 diabetes mellitus. Endocr J 2013; 60: 347– 357.
28 Liang, J, Zhang, H, Hua, B et al. Allogeneic mesenchymal stem cells transplantation in treatment of multiple sclerosis. Mult Scler 2009; 15: 644– 646.
29 Ma, L, Zhou, Z, Zhang, D et al. Immunosuppressive function of mesenchymal stem cells from human umbilical cord matrix in immune thrombocytopenia patients. Thromb Haemost 2012; 107: 937– 950.
30 Shi, M, Zhang, Z, Xu, R et al. Human mesenchymal stem cell transfusion is safe and improves liver function in acute‐on‐chronic liver failure patients. Stem Cells Translational Medicine 2012; 1: 725– 731.
31 Wu, KH, Sheu, JN, Wu, HP et al. Cotransplantation of umbilical cord‐derived mesenchymal stem cells promote hematopoietic engraftment in cord blood transplantation: A pilot study. Transplantation 2013; 95: 773– 777.
32 Wu, KH, Tsai, C, Wu, HP et al. Human application of ex vivo expanded umbilical cord‐derived mesenchymal stem cells: Enhance hematopoiesis after cord blood transplantation. Cell Transplant 2013; 22: 2041– 2051.
33 Zhang, Z, Lin, H, Shi, M et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J Gastroenterol Hepatol 2012; 27: 112– 120.
34 Kim, JH, Jo, CH, Kim, HR et al. Comparison of immunological characteristics of mesenchymal stem cells from the periodontal ligament, umbilical cord, and adipose tissue. Stem Cells Int 2018; 2018: 8429042.
35 Najar, M, Raicevic, G, Boufker, HI et al. Mesenchymal stromal cells use PGE2 to modulate activation and proliferation of lymphocyte subsets: Combined comparison of adipose tissue, Wharton’s jelly and bone marrow sources. Cell Immunol 2010; 264: 171– 179.
36 Arutyunyan, I, Elchaninov, A, Makarov, A et al. Umbilical cord as prospective source for mesenchymal stem cell‐based therapy. Stem Cells Int 2016; 2016: 6901286.
37 Siniscalco, D, Bradstreet, JJ, Sych, N et al. Mesenchymal stem cells in treating autism: Novel insights. World J Stem Cells 2014; 6: 173– 178.
38 Liu, Q, Chen, MX, Sun, L et al. Rational use of mesenchymal stem cells in the treatment of autism spectrum disorders. World J Stem Cells 2019; 11: 55– 72.
39 Ichim, TE, Solano, F, Glenn, E et al. Stem cell therapy for autism. J Transl Med 2007; 5: 30.
40 Siniscalco, D, Kannan, S, Semprun‐Hernandez, N et al. Stem cell therapy in autism: Recent insights. Stem Cells Cloning Adv Appl 2018; 11: 55– 67.
41 Sharma, A, Gokulchandran, N, Sane, H et al. Autologous bone marrow mononuclear cell therapy for autism: An open label proof of concept study. Stem Cells Int 2013; 2013: 623875.
42 Lv, YT, Zhang, Y, Liu, M et al. Transplantation of human cord blood mononuclear cells and umbilical cord‐derived mesenchymal stem cells in autism. J Transl Med 2013; 11: 196.
43 Dominici, M, Le Blanc, K, Mueller, I et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006; 8: 315– 317.
44 Geier, DA, Kern, JK, Geier, MR. A comparison of the Autism Treatment Evaluation Checklist (ATEC) and the Childhood Autism Rating Scale (CARS) for the quantitative evaluation of autism. J Mental Health Res Intell Disab 2013; 6: 255– 267.
45 Lalu, MM, McIntyre, L, Pugliese, C et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): A systematic review and meta‐analysis of clinical trials. PLoS One 2012; 7:e47559.
46 Karussis, D, Karageorgiou, C, Vaknin‐Dembinsky, A et al. Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol 2010; 67: 1187– 1194.
47 Roberts, W, Weaver, L, Brian, J et al. Repeated doses of porcine secretin in the treatment of autism: A randomized, placebo‐controlled trial. Pediatrics 2001; 107:E71.
48 Handen, BL, Johnson, CR, Lubetsky, M. Efficacy of methylphenidate among children with autism and symptoms of attention‐deficit hyperactivity disorder. J Autism Dev Disord 2000; 30: 245– 255.
49 Krupa, P, Vackova, I, Ruzicka, J et al. The effect of human mesenchymal stem cells derived from Wharton’s jelly in spinal cord injury treatment is dose‐dependent and can be facilitated by repeated application. Int J Mol Sci 2018; 19: 1503.
50 Richardson, JD, Psaltis, PJ, Frost, L et al. Incremental benefits of repeated mesenchymal stromal cell administration compared with solitary intervention after myocardial infarction. Cytotherapy 2014; 16: 460– 470.
51 Guo, Y, Wysoczynski, M, Nong, Y et al. Repeated doses of cardiac mesenchymal cells are therapeutically superior to a single dose in mice with old myocardial infarction. Basic Res Cardiol 2017; 112: 18.
52 Bolli, R. Repeated cell therapy: A paradigm shift whose time has come. Circ Res 2017; 120: 1072– 1074.
53 Wysoczynski, M, Khan, A, Bolli, R. New paradigms in cell therapy: Repeated dosing, intravenous delivery, immunomodulatory actions, and new cell types. Circ Res 2018; 123: 138– 158.
54 Jarocha, D, Milczarek, O, Wedrychowicz, A et al. Continuous improvement after multiple mesenchymal stem cell transplantations in a patient with complete spinal cord injury. Cell Transplant 2015; 24: 661– 672.
55 Ankrum, JA, Ong, JF, Karp, JM. Mesenchymal stem cells: Immune evasive, not immune privileged. Nat Biotechnol 2014; 32: 252– 260.
56 Masi, A, Lampit, A, Glozier, N et al. Predictors of placebo response in pharmacological and dietary supplement treatment trials in pediatric autism spectrum disorder: A meta‐analysis. Transl Psychiatry 2015; 5:e640.
